體內CAR-T:從載體設計到早期臨床的實操方案綱要(病毒路線) In vivo CAR科學史
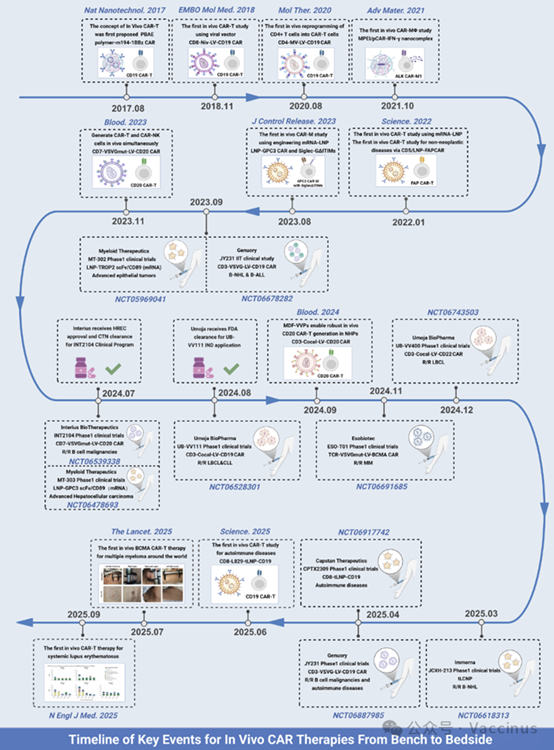
本方案提供一個從病毒載體設計到早期臨床試驗的完整閉環。
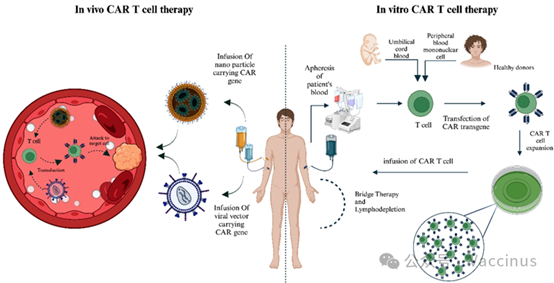
患者→(清淋or not)→靜脈注射靶向性LV載體→體內生成CAR-T細胞→治療評估
圖1:體內與體外CAR-T製造流程對比示意圖
研發全流程方案大綱:
一、 技術路線選擇與整體策略
二、 病毒載體工程化:靶向、高效、安全
三、 CAR結構設計與優化(針對實體瘤挑戰)
四、 產品表徵與功能驗證方案
五、 非人靈長類(NHP)研究及GLP毒理方案
六、 IIT(研究者發起的臨床試驗)方案框架
七、 POC驗證中的常見問題與解決方案
基於病毒載體的體內CAR-T療法研發全流程方案 (POC至IIT階段)
一、 技術路線選擇與整體策略
核心決策:
病毒載體平臺選擇,慢病毒載體(Lentiviral Vector, LV)是目前體內CAR-T工程化最成熟、臨床進展最快的平臺,其優勢在於:
- 高轉導效率:對分裂和非分裂細胞(如靜息T細胞)均有效,適合體內直接轉導。
- 穩定整合與長效表達:可實現CAR的長期、穩定表達。
- 成熟的改造與偽型技術:可通過改造包膜蛋白(Pseudotyping)實現細胞靶向。
- 臨床經驗豐富:已有多個產品進入臨床試驗(如INT2104, ESO-T01等)。
實操:以自失活(SIN)第三代慢病毒載體作為核心技術平臺,並重點開發靶向性改造的慢病毒(Targeted LV),以解決體內應用的脫靶和安全問題。
二、 病毒載體改造、生產與質控
- 病毒載體改造:降低免疫原性與提高靶向性目標:消除對非靶細胞(如肝細胞)的天然嗜性,並重靶向T細胞。
- 包膜蛋白的選擇與改造(Pseudotyping & Detargeting/Retargeting):
去靶向(Detargeting):使用突變型VSV-G(VSV-G mutant)
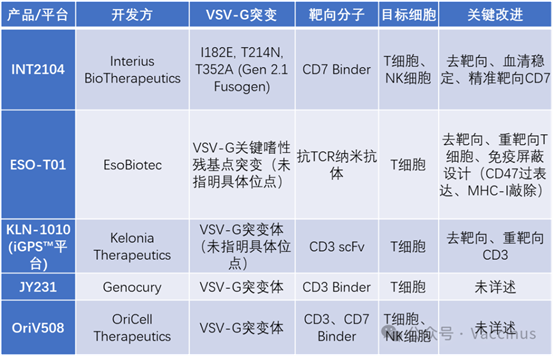
VSV-G突變技術平臺小結
除了改造VSV-G,還有其他包膜工程策略:
副粘病毒來源包膜:如Nipah病毒G蛋白、Measles病毒H蛋白、Cocal病毒包膜蛋白,其蛋白(G)和融合蛋白(F)是分離的,可以單獨對G蛋白進行去靶向和重靶向改造,而F蛋白負責膜融合,以降低其通過普遍存在的LDLR對非靶細胞的廣泛感染,工程化更靈活。
Cocal糖蛋白:與VSV-G有71.5%的同源性,也結合LDLR,但一些公司(如Umoja Biopharma)正將其用於其體內CAR-T平臺(如UB-VV111)。再靶向(Retargeting):在去靶向的包膜蛋白上融合T細胞特異性結合域。這是實現體內靶向遞送的關鍵。
- 常用靶向分子:
- 單鏈抗體(scFv):如抗CD3 scFv (OKT3, TR66)、抗CD8 scFv (Okt8opt)、抗CD4 scFv。
- DARPin(設計錨蛋白重複蛋白):如抗CD8 DARPin (MSE10)、抗CD4 DARPin 。其體積小、穩定性好。
- 雙特異性抗體(tandem Fab):如抗SINV E2 × CD3 tandem Fab。
- 連接模式:可採用串聯(Tandem)或獨立表達(Independently expression)模式將靶向分子展示在病毒顆粒表面。
- 載體骨架優化:
o自失活(SIN)設計:刪除3‘LTR的啟動子區(如U3區),防止病毒啟動子啟動下游原癌基因,是必須的安全特性。
o使用強效且相對特異的啟動子:如EF1α、PGK,或使用T細胞特異性啟動子(如CD3 promoter)以進一步限制CAR在非T細胞中的表達。
o考慮非整合型慢病毒(NILV):對於需要暫態表達或擔心插入突變的應用,可探索使用整合酶缺陷型慢病毒,但其表達持續時間較短。
- 病毒載體生產工藝優化
目標:建立穩定、高滴度、符合GMP導向的病毒生產體系。
- 生產系統選擇:
- 暫態轉染系統:使用HEK 293T/17細胞,通過多質粒(通常為3質粒或4質粒系統:包裝質粒、包膜質粒、轉移質粒(含CAR基因))共轉染。這是最靈活、最常用的臨床前和早期臨床方法。
- 穩定生產細胞系:如建立基於PG13(用於γ-逆轉錄病毒)或誘導型包裝細胞系。這有利於大規模、批次間一致的生產,但構建複雜。
- 推薦路徑:POC階段採用暫態轉染(HEK 293T),便於快速反覆運算載體設計;進入臨床申報時,需建立主細胞庫(MCB)和工作細胞庫(WCB),並考慮向穩定生產細胞系或更可控的放大工藝,如生物反應器過渡。
- 提高病毒滴度的關鍵工藝參數:
-
- 質粒比例與轉染試劑:優化包裝質粒、包膜質粒、轉移質粒的比例(如:30μg:10μg:10μg:10μg)。比較PEI、磷酸鈣、脂質體等轉染試劑的效率。
- 細胞狀態與密度:轉染時細胞應處於對數生長期,融合度約60-80%。
- 收穫時間:通常在轉染後48-72小時多次收穫上清,以獲取最高總產量。
- 濃縮與純化:臨床級產品必須進行純化。採用超速離心或切向流過濾(TFF)結合層析(如離子交換、分子排阻) 進行濃縮和去雜質。注意控制流速和壓力,避免損傷病毒顆粒。
- 無/低血清工藝:為降低牛源性雜質風險,應開發無血清或化學成分限定(chemical defined)的培養基。
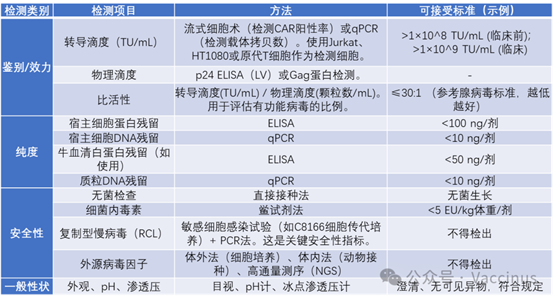
- 病毒載體品質控制(QC)與放行
目標:建立全面的品質控制策略
關鍵放行檢測項目:
三、 CAR結構設計與優化(針對實體瘤)
目標:克服腫瘤微環境抑制、抗原異質性和靶向/脫靶毒性等挑戰。
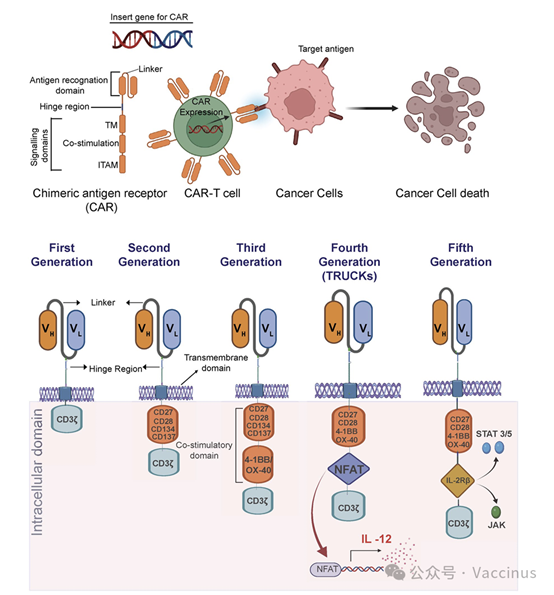
靶點選擇與驗證:選擇在腫瘤細胞上高表達、在關鍵正常組織上低或不表達的抗原。進行嚴格的組織交叉反應(Tissue Cross-Reactivity, TCR) 分析。
scFv優化:篩選高親和力、高特異性的scFv。注意親和力與抗腫瘤活性、脫靶風險的平衡(“親和力窗口”)。鉸鏈/跨膜區優化:
-
- 鉸鏈區:長度和靈活性影響CAR的空間可及性和信號傳導。常用CD8α或IgG衍生的鉸鏈。
- 跨膜區:常用CD8α或CD28的跨膜區,主要影響CAR的穩定性和與內源性信號分子的相互作用。
- 共刺激信號域組合:第二代CAR(1個共刺激域)是基礎。
- CD28域:提供強效的早期啟動信號,但可能導致T細胞耗竭。
- 4-1BB域:提供持久的存活信號,增強體內持久性,但初始啟動可能較弱。
- 實體瘤推薦:可考慮4-1BB以增強持久性,或開發雙共刺激域(如CD28-4-1BB) 的第三代CAR,但需注意信號強度調節。
- 也可其他共刺激分子,從頭篩選與改造
應對實體瘤微環境的策略(“裝甲化”CAR):
-
- 分泌細胞因數:設計CAR-T細胞共表達IL-12、IL-15、IL-7等,以抵抗微環境抑制、促進增殖。
- 表達趨化因數受體:如CXCR2、CCR4等,幫助CAR-T細胞浸潤至腫瘤部位。
- 敲除抑制性受體:在載體中整合CRISPR元件,或使用雙載體策略,同時敲除PD-1、TIGIT等抑制性受體。
邏輯門控(Logic-gated)CAR:AND,OR,NOR等邏輯門控;如通過“及閘”(AND-gate)設計,要求兩個腫瘤相關抗原同時存在才啟動,極大提高安全性。
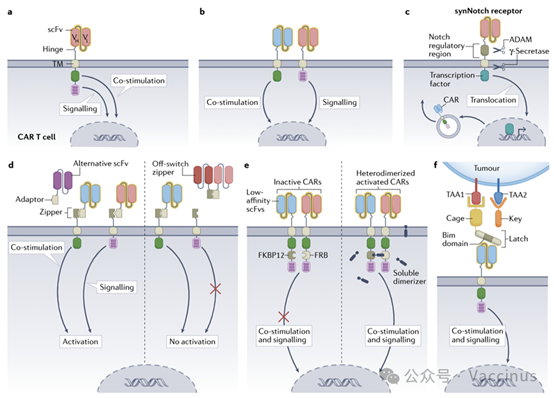
邏輯門控策略
- 可調控開關:整合藥物(如雷帕黴素)或小分子誘導的基因開關,實現CAR活性的可控開啟/關閉。
針對on target, off-tumor的策略:
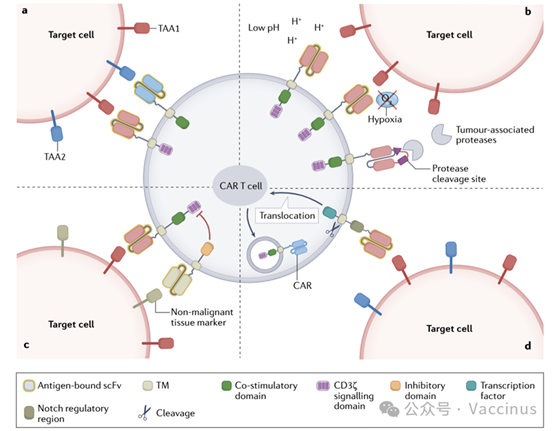
四、 體內外功能驗證方案
- 體外驗證
- CAR表達驗證:用靶向性LV轉導原代人T細胞(或PBMC),通過流式細胞術檢測CAR陽性率、平均螢光強度(MFI)和表型(CD4/CD8比例,記憶亞群)。
- 抗原特異性啟動:與表達靶抗原的腫瘤細胞共培養,檢測:
- 細胞因數釋放:ELISA檢測IFN-γ,IL-2, TNF-α。
- 脫顆粒:流式檢測CD107a等。
- 增殖能力:CFSE或Cell Trace Violet染色。
- 體外殺傷實驗:
- 即時細胞分析(RTCA):即時監測腫瘤細胞裂解(Incucyte等)。
- 標準鉻釋放實驗(目前基本淘汰):在不同效靶比(E:T)下,評估特異性殺傷效率和動力學。
- 耗竭模型:與腫瘤細胞長期反復共培養,評估CAR-T細胞的持久性和功能耗竭情況。
- 體內驗證(小鼠模型)
- 模型建立:
- 血液瘤模型:NSG或NOG小鼠,尾靜脈注射人源化腫瘤細胞系(如Raji, Nalm6)建立白血病/淋巴瘤模型。
- 實體瘤模型:NSG小鼠,皮下或原位接種表達人靶抗原的腫瘤細胞系。
- 人源化小鼠模型:向NSG小鼠移植人CD34+造血幹細胞或PBMC,建立更接近人體免疫環境的模型,用於評估體內CAR-T生成、擴增和抗腫瘤活性。
- 給藥與評估:
- 給藥:尾靜脈注射靶向性LV載體(劑量參考,如2×10^6 – 4×10^10 TU/只)。
- 監測:
- 腫瘤生長:卡尺測量(實體瘤)或活體成像。
- 體內CAR-T生成:定期采血,流式檢測人CD3+CAR+細胞的比例和絕對數量。
- 細胞因數風暴(CRS)模擬:監測血清中人源細胞因數(如IL-6, IFN-γ)水準。
- 生物分佈:實驗終點取各器官(脾、肝、肺、骨髓、腫瘤),分析CAR-T細胞的浸潤和脫靶情況。
- 長期安全性:觀察小鼠體重、活動狀態,實驗終點進行全面的組織病理學檢查。
五、 非人靈長類(NHP)實驗與GLP毒理方案
- NHP藥效/藥代動力學(PK/PD)研究
- 目的:在更接近人類的生理和免疫系統中,驗證載體的靶向性、CAR-T生成效率、持久性、抗腫瘤活性(若建立NHP腫瘤模型)和初步安全性。
- 動物:食蟹猴或恒河猴。
- 方案:
- 劑量探索:設置低、中、高三個劑量組(基於小鼠資料換算),每組n≥3。
- 給藥:單次或多次靜脈輸注靶向性LV。
- 監測:
- PK:定期采血,用qPCR定量載體基因組(VCN)在血液和各組織中的分佈與清除動力學。
- PD:流式監測CAR-T細胞(CD3+CAR+)在血液中的生成、擴增峰、表型變化和持久性(至少3-6個月)。
- 免疫原性:檢測抗載體中和抗體(NAb)的產生及其對再次給藥的潛在影響。
- 靶向/脫靶分析:分析CAR-T細胞在淋巴組織(淋巴結、脾臟)、骨髓及可能脫靶器官(如肝臟)中的分佈。
- 安全性:全程監測臨床體征、體重、體溫、血液學、臨床生化、凝血功能、細胞因數(CRS相關)。
- GLP毒理學研究
- 目的:為首次人體試驗(IIT)提供正式的安全性支援資料。
- 核心原則:遵循ICH指導原則,在GLP認證的機構進行。
- 設計:
- 動物種屬:選擇與人體生物學反應最相關的種屬,通常為食蟹猴。
- 劑量設計:設置一個無毒性反應劑量(NOAEL)、一個預計臨床等效劑量和一個明顯毒性劑量。
- 給藥途徑與週期:與臨床計畫一致(靜脈輸注)。通常包括單次給藥毒性研究和/或重複給藥毒性研究(如每週一次,連續4周)。
- 對照組:載體緩衝液對照組。
- 終點與分析:
- 全面毒代動力學(TK)。
- 全面的臨床病理學(血液學、血清生化、凝血、尿檢)。
- 免疫毒性評估:淋巴細胞亞群分析、免疫球蛋白水準、抗藥抗體(ADA)檢測。
- 組織病理學:對全部主要器官和組織進行詳細的宏觀和微觀檢查,特別關注淋巴組織、生殖腺(評估生殖細胞轉導風險)、肝臟和可能脫靶的器官。
- 生殖毒性:通常I期臨床不要求,但需評估載體在生殖腺的分佈。
- 致瘤性:長期研究(如6-12個月)觀察有無克隆性增殖或腫瘤發生跡象。
六、 IIT(研究者發起的臨床試驗)方案框架
- 試驗題目:評估XXXX,一種靶向YY的體內CAR-T細胞療法,在復發/難治性ZZZ患者中的安全性、耐受性、藥代動力學和初步療效的I期臨床研究。
- 研究設計:開放標籤、單臂、劑量遞增研究(如3+3設計)。
- 患者人群:標準治療失敗、經病理確診的ZZZ成年患者。
- 給藥方案:
預處理:根據載體特性,決定是否需要進行淋巴清除化療(如氟達拉濱/環磷醯胺)。體內CAR-T的優勢之一是可能無需強淋巴清除。
研究藥物:單次靜脈輸注XXXX(靶向性LV載體)。
劑量組:設置3-4個劑量遞增組別。
- 終點:
主要終點:安全性(不良事件AE、嚴重不良事件SAE發生率,特別關注CRS、ICANS、HLH、載體相關毒性)。
次要終點:
PK:血液中載體基因組拷貝數動態變化。
PD:外周血中CAR-T細胞比例、數量、表型及持久性。
免疫原性:抗載體中和抗體(NAb)產生。
初步療效:客觀緩解率(ORR)、緩解持續時間(DOR)、無進展生存期(PFS)。
- 關鍵監測:
CRS/ICANS管理:制定詳細的托珠單抗和糖皮質激素使用預案。
長期隨訪:根據FDA指南,對使用整合型載體的患者進行長期隨訪(至少15年),監測遲發性不良反應,特別是繼發性腫瘤。
七、 POC驗證中的常見問題與Troubleshooting
