中國CDE出臺技術指南 進一步鼓勵境外臨床試驗資料用於註冊申請
在ICH資料互認的語境下,國家藥監局進一步鼓勵境外原研藥上市及中國參與全球同步開發,這不但有利於製藥外企將更多新藥帶到中國,也利於本土企業利用審評機構對境外資料的認可,加快國外項目的引進。
2018年,國家藥監局藥品審評中心(CDE)曾在《接受藥品境外臨床試驗資料的技術指導原則》中開宗明義提及“越來越多跨國公司和國內企業通過開展國際多中心臨床試驗用於支援全球的註冊申請。CDE旨在為境外臨床試驗資料用於在我國進行藥品註冊申請提供參考,以鼓勵藥品的境內外同步研發。”時隔兩年後,CDE在今年10月發佈了《境外已上市境內未上市藥品臨床技術要求》(下稱《技術要求》)。
業內專家認為,結合兩個指導原則來看,在ICH資料互認的語境下,國家藥監局進一步鼓勵境外原研藥上市及中國參與全球同步開發,這不但有利於製藥外企將更多新藥帶到中國,也利於本土企業利用審評機構對境外資料的認可,加快國外項目的引進。
對境內外企業的影響
泰格醫藥法規事務副總裁常建青認為,《技術要求》中臨床評價按照臨床需求、有效性和安全性評價、種族敏感性分析、基於中國患者獲益/風險決策來評估,邏輯清晰,臨床試驗要求明確。“實際上要具體問題具體分析,境外企業是按照1.1類還是5.1類藥物進入中國需要重點考慮。”常建青說。
受訪外企專家認為,註冊法規條例的系統性調整,增加了進口新藥在國內外同步上市的可行性。這些新政優化了進口藥的註冊申報程式,接收境外臨床試驗資料表示中國加入ICH成員國之後和全球新藥研發的接軌成為現實。對跨國藥企總部而言,因為新藥上市速度的加快,中國市場的重要性會因此提升;對中國研發團隊來說,爭取儘早加入國際多中心臨床試驗的動力更強。
根據戊戌資料提供的資料來看,近幾年來進口藥和全球多中心臨床試驗數量明顯加大。
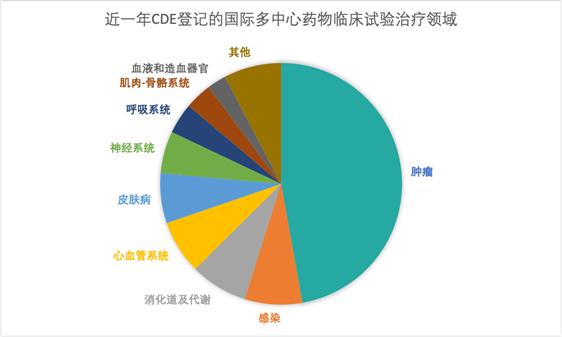
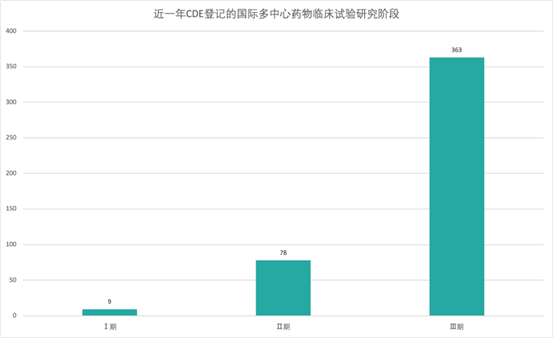
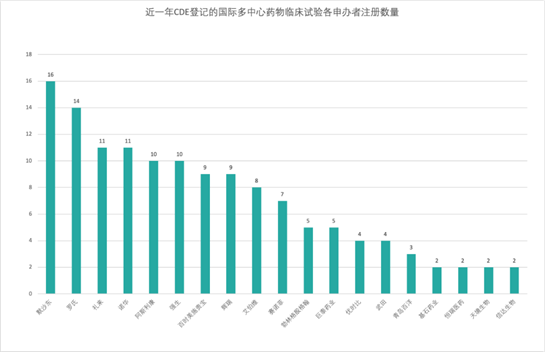
勃林格殷格翰(BI)大中華區醫學和臨床研發負責人張維博士說:“隨著一系列鼓勵同步開發和接受境外資料的技術指導原則的出臺,外企創新藥中國上市與全球有‘時間差’會逐步成為歷史。近兩年,BI啟動了中國納入(China IN)和中國關鍵(China Key)專案,中國已默認參加BI全球早期臨床試驗和註冊試驗。我們積累了一些全球同步遞交和同步審批的經驗。今年6月BI中國有一個適應症獲批僅僅比歐盟晚1.5個月;10月底,我們另一個適應症的遞交計畫僅晚於美國4天。”
什麼情況可以豁免臨床?

為了縮小國內外藥品上市時間差,一些加快上市速度的路徑尤其得到企業的關注,比如豁免臨床。《技術要求》對豁免臨床做出了說明:“藥品安全有效且無種族敏感性的,可考慮豁免境內臨床試驗”(見上圖)。今年出臺的新《藥品註冊管理辦法》中,特別提到幾個上市路徑,其中一條即為“經申請人評估無需或不能開展藥物臨床試驗,符合豁免藥物臨床試驗的,申請人可以直接提出藥品上市許可申請的路徑。”
禮來中國藥物研發及醫學事務負責人王莉博士認為:“期望在中國人群裡不開展任何臨床研究就直接在中國註冊上市,這在實操層面上並不太可能。根據ICH E5的要求,CDE明確要求企業方在遞交註冊申請時必須提供PK/PD、藥物療效和安全性的種族敏感性分析結果。該政策鼓勵境外原研藥品自臨床早期研發階段即在中國同步開展臨床試驗。這將獲得種族因素影響相關的完整證據鏈的直接證據。”
對於豁免臨床,日本製藥企業協和麒麟製藥中國研發負責人丁鉲博士有最直接的感受。他接受研發客採訪時說,該公司治療X連鎖低磷佝僂病的布羅梭尤單抗、治療斑塊型銀屑病的布羅利尤單抗被納入CDE《48個境外已上市臨床急需新藥名單》;此外,用於皮膚T細胞淋巴瘤的Mogamulizumab也經CDE評估後認為安全有效且無種族敏感性,同意豁免臨床試驗。因此,這3個品種的中國上市時間均縮短至少3年。
丁鉲說,CDE接受境外資料越來越靈活。對於境外已上市的治療罕見病、危重疾病和/或兒童患者且臨床無有效治療手段的藥物,即便沒有中國人的臨床資料,如果現階段資料提示歐美人群與日本人人種無明顯差異,就可以爭取與CDE溝通豁免臨床研究,直接申請上市。
中國藥科大學楊勁教授認為,可以借鑒美國FDA“以病人為中心的審評理念”。國外大量罕見病藥物臨床研究入組病例少,在無藥可用的情況下,以病人的最大利益為訴求,充分借鑒寶貴的資料,保證在中國病人身上獲益大於風險,應儘快豁免臨床試驗。“即便會產生種族敏感性,但因此要求它在中國進行橋接研究並不現實。”楊勁教授說。
對於《技術要求》,百濟神州全球研究及亞太臨床開發負責人、高級研發副總裁汪來博士認為十分具有指導意義,但挑戰在於審評機構如何公平、公正、可持續、一致地執行。“什麼情況豁免臨床,什麼情況開展臨床,每個藥物千差萬別。因此,審評人員對新藥風險獲益評估和把控的能力變得非常重要。”汪來博士說。
王莉博士認為,只有在用於嚴重或危及生命疾病、罕見病且無有效治療手段的藥品,或用於此類疾病且較現有治療手段具有明顯提高療效或安全性等優勢的藥品,可考慮在嚴格風險控制的前提下豁免臨床批准上市,並應開展上市後有效性和安全性臨床試驗以支持藥品全生命週期獲益/風險評估。
橋接試驗策略和資料互認的重要性

《技術要求》中對於臨床試驗的要求特別寫到,“當境外已上市的藥物安全有效但缺乏種族敏感性資料或存在種族敏感性時候,經評估,該藥品安全有效但缺乏種族敏感性資料或已有資料提示存在種族敏感性的,應開展相關橋接臨床試驗”。
據謀思醫藥聯合創始人、CEO楊見松介紹,藥企通常採用橋接試驗和國際多中心試驗;是否存在種族差異,成為藥物全球開發策略的重要考慮因素。藥物相關的種族差異包括藥代動力學、藥效學、安全性等幾個方面。據美國和日本的文獻報導,大約有20%~30%的藥物存在明顯的種族差異,需要劑量調整。
近些年來,國家藥監局頻繁在技術指南中提到 “橋接試驗”,ICH E5即是所稱的橋接試驗,即在新地區進行附加的小規模研究並提供新地區藥動學、有效性、安全性的臨床資料以證明沒有種族差異,使得國外臨床資料能外推至新地區人群。
亞洲的橋接試驗始於日本,中國工程院院士桑國衛早在上世紀90年代就撰文大力提倡運用橋接試驗引入國外新藥臨床試驗。中國加入ICH之後,對於橋接試驗的認識和運用也逐漸加大。
南京醫科大學流行病與衛生統計系教授辛衛權等學者曾在《新藥臨床試驗中的橋接試驗》中撰文寫道,橋接策略應該包含在企業全球研發計畫之中,如,建議製藥企業和藥品管理部門共同討論,在原地域的Ⅰ、Ⅱ、Ⅲ期臨床試驗中加以考慮。見下表:
縱覽日本新藥申請中成功使用橋接試驗的藥品,使用橋接策略可分四種模式。一,以健康受試者為物件的,獨立的PK研究和劑量-反應的臨床試驗;二,以健康受試者和患者為物件的,獨立的PK研究和Ⅱ期劑量-反應的臨床試驗;三,不進行獨立的PK研究,而在臨床試驗中包含PK研究;四,進行獨立的PK研究,而且在臨床試驗中也包含PK研究。
據原外商製藥協會(RDPAC)中國總裁、原安斯泰來中國公司董事長兼總經理卓永清回憶說,他擔任安斯泰來總經理時,中國還沒有加入ICH,藥監局要求進口藥做200對以上病例的臨床試驗,沒有使用橋接試驗,現在加入ICH之後,監管和學術界加大討論橋接試驗。他認為,我國早日接受並實施橋接試驗意義重大而深遠,希望行業、學術界以及監管部門充分溝通,早日引進橋接試驗的實施,推動進口創新藥及時進入我國,供醫療人員用於需要的病患。
汪來博士以澤布替尼在美國上市為例說明了資料互認的重要性。他說,FDA之所以快速接受澤布替尼的試驗資料是基於以下兩點:第一,澤布替尼申請B細胞淋巴瘤適應症時,FDA在經過依布替尼治療套細胞淋巴瘤的評審後,對澤布替尼的安全性資料心中已有把握。第二,FDA是基於兩項臨床試驗的有效性資料作出批准,一項是單臂多中心的Ⅱ期中國臨床試驗BGB-3111-206(NCT03206970),另一項是在澳大利亞開展的國際Ⅰ/期BGB-3111-AU-003(NCT02343120)試驗,“澳大利亞的試驗入組了許多例白種人患者,該國患者資料對FDA來說也可作為白種人試驗資料的來源。”汪來博士說,FDA對種族差異也非常重視,但不限於一定是美國境內受試者的資料。CDE在考慮中國人種資料時,或可參考FDA的做法,只要是亞洲人種資料都能依據ICH E5資料互認的科學準則來接納,而不一定完全來自於中國境內的亞洲患者資料。楊勁教授也認為,亞洲各國之間,如中國、日本和韓國要儘早實現資料互認,藥監機構大力合作或迎來亞洲創新藥發展的機遇。
境內企業引入項目獲益多
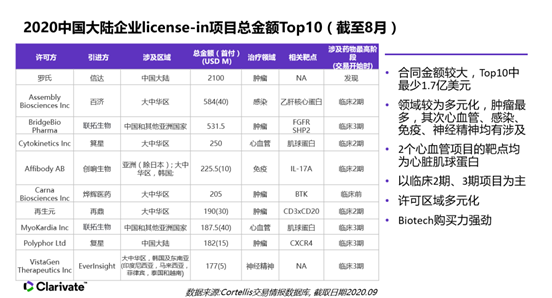
我國引入境外的品種數量也逐漸增多。科睿唯安生物科學製藥事業部解決方案顧問鮑書馨女士在日前廣州召開的 “海外新藥引進項目”高峰論壇上,介紹了2020年創新藥重大合作案例,用資料分析了截至2020年8月最熱門的引進項目及領域。雖然腫瘤依然是熱點,但是由於疫情影響,藥品交易品種正在悄然發生變化。對於小規模藥企來說,尋找最熱門的項目才是最優的選擇。
初創公司同樣獲益良多。和其瑞醫藥去年從拜耳公司獲得在全球開發與產業化靶向泌乳素(PRL)受體單克隆抗體(HMI-115)的權利。公司創始人婁實博士說,HMI-115抗體,作為全球首創(FIC)新藥,研發和產業化的最佳路徑必定是全球同步。
“我們一開始,就制定了全球研發計畫,已完成符合ICH及EMA、FDA、和NMPA等主要法規地區要求的臨床試驗用藥CMC研究和臨床批生產。HMI-115早已在中國和德國已完成了一系列臨床前藥理毒理試驗和Ⅰ期臨床試驗,顯示出突出並持久的藥效和良好的耐受性和安全性。該產品全球權益轉讓給和其瑞之後,我們和眾多臨床醫生都期待早日在歐洲、美國和中國就多個適應症能夠同步開啟多中心Ⅱ期臨床研究。我們很高興看到《技術要求》的發佈。它對加速我們在中國開展橋接試驗和Ⅱ、Ⅲ期臨床試驗很有幫助。”婁實博士說。
而領晟醫藥作為一家擅長引入歐洲創新藥項目的公司,最早也經歷了國家藥監局面對歐洲人種的試驗資料不知如何評估的局面。創始人董事長宋燕博士說,領晟2014年開始做項目引進時,國內臨床審批很難,那時領晟選擇項目採取品質優先,關注產品的市場定位、創新程度、在其適應症領域的特點、項目落地能力等,這些都與行業環境及政策息息相關,而近幾年政策調整極快,“根據有效性安全性和是否種族差異性作為原則,我們加強與藥審中心溝通後,CDE接受了境外的試驗資料。領晟申報的專案至今沒有被退回或被要求補充資料的。”宋燕說。
接受境外資料的監管過程
中國藥監接受境外臨床試驗資料經歷了一個漫長的過程。2007年的《藥品註冊管理辦法》規定,臨床試驗用藥物應是在境外註冊或已進入Ⅱ期、Ⅲ期臨床試驗藥物,國外早期臨床試驗資料並不能直接拿到中國來申報。
2015年,原國家藥監總局發佈了《關於發佈國際多中心藥物臨床試驗指南(試行)的通告》,該指導原則要求對全球臨床試驗資料分析上,針對亞洲人群的有效性和安全性與非亞洲人群進行比較和趨勢性分析;還要針對我國人群的有效性和安全性資料與其他國家人群的相應資料進行比較和趨勢性分析,加大了對國際多中心臨床資料的認可和評估。
隨後在2017年10月8日,國務院辦公廳印發了《關於深化審評審批制度改革鼓勵藥品醫療器械創新的意見》中,第六條為:接受境外臨床試驗資料。在隨後2017年10月10日,原CFDA公佈了《關於調整進口藥品註冊管理有關事項的規定》,對進口藥品註冊要求進行了調整:在中國進行國際多中心臨床試驗,允許同步開展Ⅰ期臨床,取消臨床試驗用藥物應當在境外註冊,或已進入Ⅱ期或Ⅲ期臨床試驗的要求;對以國際多中心臨床試驗資料提出免做進口藥品臨床試驗的註冊申請,符合《藥品註冊管理辦法》及相關檔要求的,可以直接批准進口。
2018年,CDE兩次發佈了《關於徵求境外已上市臨床急需新藥名單意見的通知》,遴選了境外已上市臨床急需新藥名單,可提交或補交境外全部研究資料和不存在人種差異的支持性材料,直接提出上市申請。2019年,CDE出臺《臨床急需藥品附條件批准上市技術指導原則(徵求意見稿)》,對於治療罕見病的臨床急需藥品,境外已批准上市或境內已有臨床資料顯示其療效並能預測其臨床獲益的都可以基於替代終點、中間臨床終點或早期試驗資料而批准上市,能提早應用于急需的病人。
“眼下製藥企業都以美國作為第一上市目標,將美國和其他國家的新藥儘快引入中國;同時讓中國成為全球多中心臨床試驗的首選國之一,讓中國患者享受全球藥物研發的同步進展,是國家首要的任務。”楊勁教授說,加快境外急需藥物在中國上市因一部大眾電影《我不是藥神》的上映,而變得刻不容緩。
總體而言,正如《技術要求》中寫道,無論對於監管還是企業,分析申報適應症在我國的疾病流行病學現狀、疾病嚴重程度和預後,如現有治療手段有其局限性,要明確該藥品與國內治療手段的優勢。對用於臨床缺乏有效治療手段的危重疾病和罕見病治療藥品等,監管機構持鼓勵態度,境內外製藥企業也將有大發展,最終滿足全球的患者。