中國細胞治療監管解讀和上市前合規策略
中國細胞治療監管解讀和上市前合規策略 2022-08-15 20:37 發表於北京
根據Nature的一項統計顯示,2021年中國細胞治療研發管線數量達到了695種,和美國一起主導了全球細胞治療研發管線[2] 。在細胞治療監管領域,中國監管部門陸續出臺的細胞治療相關政策制度,也逐漸構建起了中國特有的細胞治療領域監管體系。
細胞藥物代表產品和企業▉ 細胞治療分類
目前,細胞治療分為免疫細胞治療與幹細胞治療兩種。其中,免疫細胞治療又主要分為以下幾類:

細胞藥物代表產品和企業近年來,中國細胞治療領域的研究主要集中在CAR-T細胞療法。值得注意的是,在全球8款獲批的CAR-T療法中有兩款產品來自中國,分別是:2021年6月獲批上市的阿基侖賽注射液(複星凱特產品)和2021年9月上市的瑞基奧侖賽注射液(藥明巨諾產品)。然而,TCR-T療法和幹細胞藥物,目前我國處於早期發展階段,暫無獲批上市的產品。
細胞治療法規概述▉ 中國細胞治療監管體系概述
經梳理,中國的細胞治療監管分為醫療技術和藥品兩個路徑,大致經過了三個階段,分別是1993年至2015年,監管較為寬鬆;2016年,嚴格調整階段;2017至今的全面規範階段,具體內容如下:
(一)1993年-2015年較為寬鬆階段

(二)2016年,嚴格調整階段
2016年以前,我國已認識到細胞治療的廣闊前景,各項政策以強調細胞治療的重要為主,監管總體寬鬆,細胞治療企業處於自由發展的狀態,大量開展臨床研究專案,也出現了不少細胞治療亂象,尤其是2016年4月的“魏則西”事件,反映出當時我國細胞治療存在監管漏洞,隨後,國家調整了相應的監管尺度。

(三)2017年後,全面規範階段
國家各部委和地方政府於2017年開始出臺相應檔,大力推動細胞治療技術研究及應用,規範和引導整個細胞治療產業的向前發展。


值得注意的是,2021年以來,中國多個省/市發佈了相關檔(見下表)支援細胞治療產業良性發展和促進細胞治療技術研發的國際合作及引進等,可以預見,未來細胞治療領域的外資准入條件、發展監管將呈現出放寬趨勢。

合規監管要點和合規建議
外資限制合規監管要點和合規建議
中國《鼓勵外商投資產業目錄(2020年版)》將細胞治療藥物研發與生產(禁止外商投資領域除外)列為外商投資鼓勵目錄。人體幹細胞、基因診斷與治療技術開發和應用在2022年1月1日起施行的《外商投資准入特別管理措施(負面清單)(2021年版)》中被列為禁止外商投資領域。值得注意的是,根據《基因修飾細胞治療產品非臨床研究與評價技術指導原則(試行)(徵求意見稿)》與《免疫細胞治療產品藥學研究與評價技術指導原則(徵求意見稿)》的規定,CAR-T細胞治療和TCR-T細胞治療屬於基因修飾的免疫細胞治療,而非幹細胞治療。因此,目前在我國,外商可投資CAR-T治療和TCR-治療等免疫細胞治療,而不能直接投資幹細胞治療。
實踐中,根據《外商投資法》第36條等相關規定,外籍股東所持有的公司股權存在被商務主管部門限期處置的風險[11] 。這種情況,通常公司會對其內部架構進行設置與調整。其中,常見的安排方式是:第一步,先另設一家內資公司作為細胞治療產品研發等實體;第二步,相關主體和內資公司簽署《獨家業務合作協定》《獨家購買權協定》《股權質押協定》及《授權委託書》等一系列控制協定(“控制協定”或“VIE”);第三步,將完成內資公司股權質押的工商登記作為外商投資交割後一定期限內義務。需要注意的是,前述實踐安排方式的可行性、合法性、有效性取決於未來法律法規的不時更新要求和相關政府部門的監管口徑。考慮到,實踐中具體的監管口徑將在一定程度上受制於具體監管部門的酌情裁量,在瞭解前述風險的基礎上,還需與相關監管部門保持溝通,以明確可能不時調整的監管要求和口徑。
細胞治療臨床研究合規監管要點及合規建議
在中國臨床研究/試驗主要分為兩種,分別是研究者發起的以提高臨床治療水準為主要目的的臨床試驗(Investigator-Initiated Clinical Trial, IIT)和醫藥企業發起的藥物臨床試驗(Industry-Sponsored Clinical Trial, IST)。前者是為了提高上市藥品的臨床治療水準或開展醫學研究等,後者是為了證明新藥物的有效性和安全性以申報藥品註冊上市。目前我國對IIT和IST分別通過不同的法律監管體系進行規制:
中國採取IIT臨床研究的,細胞治療企業應防止擅自收費、非合理使用經費等違法行為發生。否則,根據《臨床研究管理辦法》第27條的規定,衛生專業技術人員將予以相應處理;醫療機構未履行監管職責,也將承擔相應行政責任;構成犯罪的,移交司法機關處理。
中國細胞治療企業運營資質合規監管要點及合規建議
按藥品進行註冊申報和商業化,則細胞治療企業需在研發、註冊、生產、經營及使用各個環節滿足藥品對應的監管要求。根據我國《藥品管理法》的規定:
在研發環節,企業應遵守《藥物非臨床研究品質管制規範》(GLP)、《藥物臨床試驗品質管制規範》(GCP),確保研發全過程符合法律規定;在註冊環節,實行MAH制度,MAH應建立藥品品質保證體系,配備專門人員獨立負責藥品品質管制,MAH可通過簽訂委託協定和品質協定委託其他企業生產,但不得通過品質協議將法定只能由MAH履行的義務和責任委託給受託方承擔;
- 在生產環節,企業應符合《藥品生產品質管制規範》(GMP)對企業人員、廠房設施、生產環境、生產管理、品質管制等環節的要求,建立企業藥品品質管制體系,滿足藥監部門的藥品GMP條件;
- 在經營環節,無論是批發還是零售,企業應遵循《藥品經營品質管制規範》(GSP),在藥監部門規定的時間內達到GSP要求。
實踐中,違反GMP及GSP的企業不在少數,2020年末,遼寧藥監局、廣西藥監局、天津市市監委公佈了對5家藥企違反GMP的處罰公告,處罰結果為警告或暫停生產;2020年12月,湖南省藥監局公示了《湖南省藥品流通飛行檢查及處理情況通報》,在對30家經營企業的飛行檢查中,共有24家被責令限期整改,6家企業嚴重違反藥品GSP要求,被處以註銷《藥品經營許可證》、暫停經營等處罰。由此,細胞治療企業在生產經營過程中應注意GMP和GSP相應要求,以減少被行政處罰的風險。
此外,對於開展幹細胞、免疫細胞療法研究的機構及企業,應符合《幹細胞臨床研究管理辦法(試行)》及參照《體細胞治療臨床研究和轉化應用管理辦法(試行)(徵求意見稿)》的資質規定。

中國人類遺傳資源跨境傳輸合規監管要點及合規建議
中國人類遺傳資源跨境傳輸依據是2019年國務院公佈的《人類遺傳資源管理條例》(下文簡稱《條例》)和全國人大常委會於2020年公佈的《生物安全法》。這兩部檔對我國人類遺傳資源的使用、攜帶及保護等環節均明確了禁止和限制性規定。由於我國法律對人類遺傳資源的監管規定涉及採集、保藏、利用、對外提供(傳輸)等多個環節,且細胞治療企業在研發、經營過程中難免涉及監管要求,由此,不排除存在相應監管風險。特別地,《條例》中規定境外組織、個人及其設立或者實際控制的機構(外方單位)不得在我國境內採集、保藏我國人類遺傳資源,不得向境外提供我國人類遺傳資源等。具體如下:

此外,我們也關注到了監管實踐中的一些行政處罰案例:例如,科技部於2018年10月24日公佈人類遺傳資源行政處罰資訊,華大基因、藥明康得等在內的共六家企業機構被處罰,違法事實包括:

再如,百時美施貴寶(中國)投資有限公司(下文稱施貴寶)作為申辦方委託愛恩康臨床醫學研究(北京)有限公司(下文稱愛恩康)申請我國人類遺傳資源國際合作活動行政許可,愛恩康相關業務人員偽造公章和法人簽字, 向管理機構提交虛假申請材料,存在相應違規獲得中國人類遺傳資源收集(國際合作)活動行政許可的情形,科技部於2020年12月20日公佈對其做出處罰決定。

如細胞治療企業日常業務涉及到我國人類遺傳資源跨境的,在將我國人類遺傳資源材料運送、郵寄、攜帶出境時,應向科技部有關部門進行申報備案、審核。否則,根據《條例》第38條規定,將面臨由海關依照法律、行政法規的規定進行相應處罰。
中國細胞治療企業的上市審核合規監管要點及合規建議
截至2021年8月31日,我國共有五家CAR-T細胞治療企業在海外(含香港)上市,其上市的基本情況及上市結構如下:
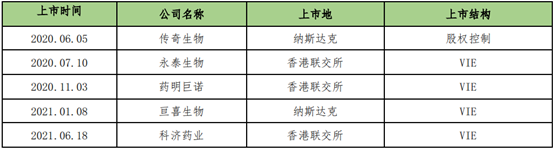
目前,暫無細胞治療企業在境內上市,為了給予企業合規建議,下面對科創板中生物醫藥企業的核心審查要求進行總結概括。根據《上海證券交易所科創板股票上市規則》(《科創板上市規則》)的規定,對於申請第五套上市標準的生物醫藥企業,至少有一項核心產品獲准開展2期臨床試驗[22] 。2021年度(截止到2021年12月29日),共有17家生物醫藥企業科創板IPO終止,經審查其終止的具體原因可以瞭解到,上市審核監管重點,主要包括以下四個方面:

由此,對於具有在境內上市目標的細胞治療企業,應重視企業的自主研發核心技術產品和誠信合規運營,保障企業各類資料的真實性等。
監管體系完善和合規問題探討
中國細胞治療監管體系存在著區分不細、“一刀切”、監管主體及對應職責不明確等問題,制約我國細胞治療行業的進一步規範發展。應明確細胞治療不同階段、不同產品的監管主體,將幹細胞及免疫細胞治療按照產品風險等級制定不同的監管規則。對於較低風險的細胞治療,可以通過醫療技術備案來簡化審批手續,加快臨床研究或試驗進程;對於高風險細胞治療,可通過審查批准程式嚴格規範其臨床研究設計,規避試驗風險。國家相關監管體系將日趨規範化、全面化和細緻化,對於我國細胞治療企業機構來說,在具有研發實力的基礎上,完善實驗室及臨床試驗品質監控,遵守註冊/備案及GMP和GCP相關要求也是細胞治療企業機構必須做好的一環。